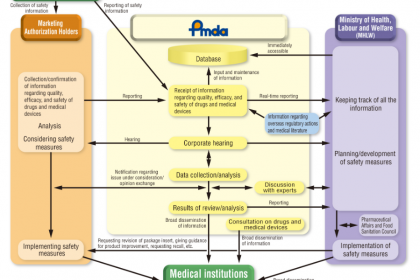
在全球醫療器械監管體系中,日本憑借嚴謹的分類標準、清晰的監管層級、完善的法律法規及專業化的審評機構,構建了覆蓋醫療器械全生命周期的監管與注冊框架。日本醫療器械的管理核心由衛生勞動和福利部(Ministry of Health, Labor and Welfare,MHLW,即日本厚生省)統籌,其管轄的獨立行政法人藥品和醫療器械綜合機構(Pharmaceuticals and Medical Devices Agency,PMDA)承擔核心技術審評、復核等職能。本文系統梳理日本醫療器械的分類標準、核心監管機構(PMDA與MHLW)職責、相關法律法規、全流程注冊要求,深入解析各環節核心要點,為醫療器械企業拓展日本市場、合規開展注冊申報及相關從業者提供、、系統的行業參考(本文所指醫療器械不含體外診斷試劑)。
一、核心監管機構解析:PMDA與MHLW的職能分工與關聯
日本藥品和醫療器械的監管體系以“MHLW統籌管控、PMDA專業執行”為核心,兩者權責清晰、協同聯動,共同保障醫療器械的、有效與合規性,明確界定了日本醫療器械監管的核心架構。
(一)厚生勞動省(MHLW):統籌監管的核心主管部門
MHLW全稱為Ministry of Health, Labor and Welfare(衛生勞動和福利部),即日本傳統意義上的厚生省,是日本醫療器械監管的主管部門,主要承擔統籌規劃、法規制定、最終審批等宏觀管控職能,具體職責包括:
- 統籌負責日本藥品、醫療器械的管理,制定監管戰略與發展規劃;
- 審批發布醫療器械相關法律法規、標準規范,明確監管要求與實施細則;
- 對PMDA的工作進行監督與指導,審定PMDA提交的醫療器械審評結果、認證結論,最終作出承認、批準等決策;
- 負責醫療器械上市后監管的宏觀統籌,協調處理重大風險與違規事件。
(二)PMDA:專業執行的技術審評機構
PMDA全稱為Pharmaceuticals and Medical Devices Agency(獨立行政法人藥品和醫療器械綜合機構),是MHLW管轄下的獨立行政法人,專注于醫療器械、藥品等的技術審評、管控等專業工作,是日本醫療器械監管體系的核心執行機構,具體職責包括:
- 醫療器械上市前技術審評,包括新器械、改良器械、仿制器械的審評復核,為MHLW的最終承認提供專業技術支撐;
- 開展醫療器械相關研究工作,完善審評標準與技術規范,參與監管體系優化;
- 承擔醫療器械上市后對策,跟蹤產品使用,處置隱患與不良事件;
- 負責健康損害救濟相關工作,規范救濟流程,保障醫療器械使用者的合法權益;
- 開展制造商注冊審核、質量管理體系審核等,規范企業生產經營行為;
- 為企業提供臨床試驗、注冊申報等相關咨詢服務,指導企業合規申報。
(三)PMDA與MHLW的核心關聯
PMDA隸屬于MHLW,接受MHLW的監督與指導,兩者形成“宏觀統籌+專業執行”的協同機制:PMDA負責所有醫療器械的技術復核、審評、審核等專業工作,提出審評意見與建議;MHLW基于PMDA的專業意見,負責最終的審批、承認、法規發布等宏觀管控,確保監管工作的專業性與性統一。
二、日本醫療器械相關法律法規體系
日本藥品、醫療器械管理法律法規體系層次清晰,分為法律、政令/法令、告示/省令三個層級,層層銜接、明確具體,為醫療器械分類、監管、注冊等工作提供了明確的法律依據,確保監管工作有法可依、有章可循。
1. 法律:由日本議會批準通過,是醫療器械監管的層級法規,具有法律效力,明確醫療器械監管的核心原則、整體框架與核心要求,指導后續政令、告示的制定。
2. 政令/法令:由日本政府內閣批準通過,是對法律的細化與補充,明確法律條款的具體實施細則,界定各相關部門、機構的權責分工,確保法律能夠有效落地執行。
3. 告示/省令:由MHLW(厚生省)大臣批準通過,是醫療器械監管的具體實施標準,涵蓋醫療器械分類基準、注冊流程、審評標準、質量管理要求等具體內容,是企業合規申報、監管機構開展工作的直接依據(如MHLW Ordinances NO.69關于質量管理體系的要求)。
其中,《藥品和醫療器械法案》(Pharmaceutical and Medical Device Act, PMD Act)是日本醫療器械監管的核心法律,明確了醫療器械制造商注冊、產品注冊、質量管理等核心要求,是所有醫療器械相關活動的根本遵循。
三、日本醫療器械分類標準:基于風險等級的分級體系(結合JMDN編碼)
日本醫療器械的分類核心遵循“風險等級適配監管強度”原則,結合日本醫療器械術語集(Japanese Medical Device Nomenclature, JMDN)編碼,明確界定器械分類與注冊登記路徑,依據產品風險等級由低到高,分為Ⅰ類(一般)、Ⅱ類(管制)、Ⅲ類(高度管制)、Ⅳ類(高度管制)四個等級,各類別界定清晰、適用范圍明確,為后續差異化監管與注冊申報提供基礎依據。
1. 一般醫療器械(Ⅰ類):風險等級,定義為“即使發生不良事件,對人體的風險也極低”的產品,如手術刀等。此類產品結構簡單、性高,無需復雜的性驗證即可保障使用,監管與注冊方式以地方政府備案為主,無實質性審查。
2. 管理醫療器械(Ⅱ類,管制醫療設備):風險等級中等,定義為“即使發生不良事件,對人體的風險也比較低”的產品,如電子內窺鏡、消化器官用導管等。此類產品結構與功能相對復雜,存在一定潛在風險,需通過第三方認證機構(RCB)負責審查,完成認證后即可合規上市。
3. 高度管理醫療器械(Ⅲ類,高度管制醫療設備):風險等級較高,定義為“在發生不良事件時,對人體的風險比較高”的產品,如透析器、人工骨骼、人工呼吸器、心臟血管用球囊導管等。此類產品技術難度較高,直接影響人體健康,需由PMDA進行專業技術審評,經MHLW(厚生省)承認后,方可上市。
4. 高度管理醫療器械(Ⅳ類,高度管制醫療設備):風險等級,定義為“對患者的侵入性高、在發生不良事件的情況下有可能直接導致生命危險”的產品,如起搏器、人工心臟、支架等。此類產品性要求,直接關系人體生命,需由PMDA進行、嚴格的技術審評,經MHLW(厚生省)承認后,方可上市。
截至2018年4月1日,日本MHLW(厚生省)已制定946個認證基準、45個承認基準和9個審查指導原則,結合JMDN編碼,為各類醫療器械的分類界定、監管實施、注冊申報提供了明確的標準依據,確保分類與監管的科學性、統一性。
四、日本醫療器械注冊全流程詳解(合規申報核心路徑)
日本醫療器械注冊流程規范、環節清晰,核心遵循“分類確定—主體任命—制造商注冊—體系審核—申報審批”的邏輯,不同風險等級產品的注冊要求有所差異,整體流程可分為四大核心步驟,確保注冊工作合規、落地。
(一)步:確定產品分類(核心前提)
產品分類是日本醫療器械注冊的核心前提,需依據JMDN編碼及風險等級,明確產品所屬類別(Ⅰ類、Ⅱ類、Ⅲ類、Ⅳ類),不同類別對應不同的注冊路徑、審查機構與監管要求。企業需先完成產品分類界定,確保后續注冊工作貼合產品特性,避免因分類偏差導致注冊延誤或違規。
(二)第二步:任命MAH/D-MAH(上市許可持有人)
根據日本醫療器械監管要求,所有類別醫療器械在開展上市前申請或審批前,必須先任命Marketing Authorized Holder(MAH,日本上市許可持有人)或Designated Marketing Authorization(D-MAH,指定上市許可持有人),負責管理產品上市申報、后續合規管控等相關事宜,具體要求如下:
- MAH:企業需拿到對應類別產品的MAH執照后,方可提出具體產品的上市申請,負責產品上市后的管控、投訴處理等;
- D-MAH:針對外國企業(在日本無辦事處),必須任命一名在日本持有營業執照的D-MAH,主要負責協調貨物放行給外國企業的經銷商,處理產品投訴、警戒信息等相關事宜,是外國企業進入日本醫療器械市場的核心銜接主體。
(三)第三步:進行制造商注冊(MR/FMR)
根據PMD Act(《藥品和醫療器械法案》)要求,醫療器械產品投放到日本市場前,制造商必須完成注冊登記,獲得相應注冊證書,該證書是提交醫療器械產品注冊申請的必備條件,具體分為兩種類型:
1. 制造商注冊(MR):適用于日本本國制造商,需向當地地方當局提交制造商注冊(MR)申請,審核通過后獲得MR證書,明確生產場所合規性;
2. 外國制造商注冊(FMR):適用于外國制造商,需向PMDA提交外國制造商注冊(FMR)申請,審核通過后獲得FMR證書,確保外國制造商的生產場所、質量管理符合日本監管要求。
MR與FMR證書均為產品注冊申報的前置條件,企業需在提出產品注冊申請前,完成制造商注冊并取得對應證書。
(四)第四步:質量管理體系審核(J-GMP/QMS)
質量管理體系合規是日本醫療器械注冊的核心要求之一,核心遵循J-GMP(日本藥品生產質量管理規范,MHLW Ordinances NO.69)及QMS(質量管理體系)相關標準,不同類別產品的審核要求差異化明確:
- Ⅰ類醫療器械:無需進行J-GMP審核,僅需完成地方政府備案即可;
- Ⅱ類醫療器械:由注冊認證機構(RCB)負責開展J-GMP審核,審核通過后,方可進入后續注冊申報環節;
- Ⅱ類(除特殊控制外)、Ⅲ類、Ⅳ類醫療器械:由PMDA負責開展QMS審核,嚴格核查企業質量管理體系的合規性、有效性,審核通過后,方可進入技術審評環節。
(五)補充說明:不同類別產品的后續申報與審批
完成上述四大核心步驟后,企業需根據產品類別,提交相應的注冊申報資料,進入最終的審評與審批環節:
- Ⅰ類醫療器械:完成地方政府備案,無實質性審查,備案完成后即可上市;
- Ⅱ類醫療器械:向RCB提交申報資料,由RCB完成審查,審查通過后即可上市;
- Ⅲ類、Ⅳ類醫療器械:向PMDA提交申報資料,由PMDA完成技術審評,審評通過后報MHLW(厚生省)承認,獲得承認后即可上市。
五、核心總結
日本醫療器械監管與注冊體系以“風險分級、管控、合規”為核心,形成了“MHLW統籌、PMDA執行、多機構協同”的完善架構。MHLW作為主管部門,負責法規制定與最終審批;PMDA作為專業執行機構,承擔技術審評、體系審核等核心工作;結合JMDN編碼的分類標準、層次清晰的法律法規,以及規范的注冊流程,構建了覆蓋醫療器械全生命周期的監管與注冊生態。
對于醫療器械企業而言,把握產品分類、明確PMDA與MHLW的職能分工、嚴格遵循注冊全流程要求,是合規進入日本市場、實現長效發展的關鍵。本文系統梳理的分類標準、監管機構、法律法規及注冊流程,可為相關企業與從業者提供的參考,助力其完成合規申報與運營。